Skupaj je 45 različnih bolezni shranjevanja lizosoma, ki so heterogena skupina prirojenih presnovnih bolezni. Ljudje, ki trpijo za katero koli od teh bolezni, imajo genetsko napako. Vse bolezni shranjevanja imajo skupno eno stvar: določen encim je odsoten ali le delno deluje.
Kaj je bolezen shranjevanja lizosoma?

© designua - stock.adobe.com
Te bolezni prirojenih shranjevanj so redke, saj jih prizadene manj kot pet na 10.000 ljudi. Različne bolezni imajo zelo različen potek, simptomi pa so lahko zelo različni.
Najbolj znane oblike bolezni shranjevanja lizosoma so Fabryjeva bolezen, Gaucherjeva bolezen, Pompejeva bolezen in mukopolisaharidoza (MPS). Pogosto jih imenujejo "sirote medicine", ker je pot do točno določene diagnoze in primerne terapije lahko zelo dolga. Včasih lahko trajajo leta, da prizadeti ugotovijo, kaj se z njimi dogaja.
vzroki
Za bolezni shranjevanja lizosoma so značilne nekatere oblike dednih presnovnih bolezni. Pacientu primanjkuje pomembnega encima, ki zagotavlja nemoteno delovanje metaboličnega ravnovesja. V manj izraziti obliki ta encim vsaj ni prisoten v zadostnih količinah.
Naloga encimov je odstranjevanje onesnaževal in odpadnih snovi, ki se v človeškem organizmu naberejo prek metabolizma prek lizosomov, ali ponovno obdelati tako, da se simptomi ne pojavijo.
Če pride do pomanjkanja encimov, ta nemoteno delujoč cikel odstranjevanja ni več zagotovljen. Škodljive snovi se naselijo v celicah in motijo presnovni cikel. V začetni fazi motnje nimajo opaznega učinka, obstaja le nekaj omejitev. Če pa presnovna motnja zaradi pomanjkanja encima ostane nezdravljena, se simptomi množijo, ker celice postanejo zelo razširjene.
Simptomi, tegobe in znaki
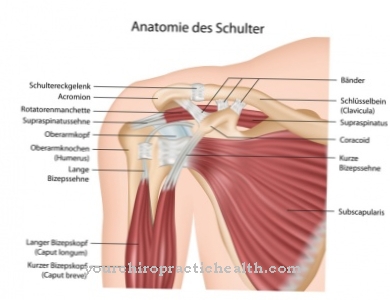
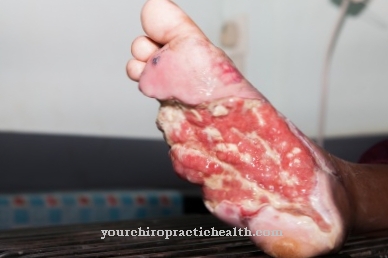
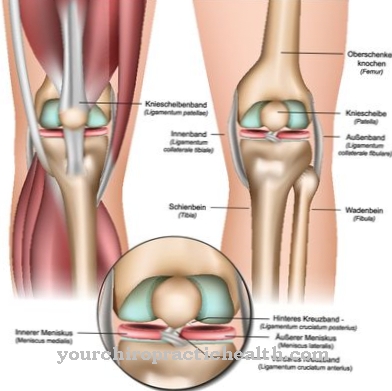
V najslabšem primeru te gredo pod. Posledice so poškodbe kosti, živčnega sistema, vranice, ledvic, mišic ali srca. Zaradi zmanjšane ali odsotne encimske aktivnosti Fabryjeva bolezen povzroči, da se maščoba (globotriaosilceramid, Gb3) shrani v celicah. Te neželene usedline lahko privedejo do močnih bolečin v prstih ali prstih, možganske kapi in okvare ledvic.
Diagnoza in potek bolezni
Ta klinična slika hkrati vpliva na različne sisteme: krvne žile, ledvice, srce in živčni sistem. Avtosomska recesivna Gaucherjeva bolezen povzroči mutacijo encima "beta-glukocerebrosidaza" in vodi do kopičenja substrata znotraj celic, zlasti v makrofagih (fagocitih), ki spadajo v retikulo-endotelni sistem. Krvna slika se spreminja, jetra in vranica se povečajo, kosti pa bolijo.
Bolezen je progresivna in je večinoma etnična, saj se v večini primerov pojavi pri ljudeh judovskega porekla. Pompeška bolezen je znana tudi kot "pomanjkanje kisle maltaze". Klinična slika spada v skupino glikogeneze tipa II. Prizadene osebe nimajo encima "alfa-1,4-glukozidaza" (kisla maltaza) ali pa ni na voljo v zadostnih količinah. Zaradi motenega razpada glikogena v mišicah bolniki trpijo zaradi uničenja mišičnih celic v obliki shrambe sladkorja.
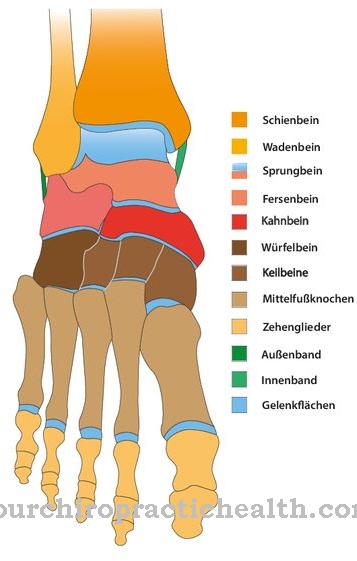
Mukopolisaharidoza tipa I (MPS), znana tudi kot Hunterjeva bolezen, ima različne klinične vzroke. Hurlerjeva bolezen je najtežja oblika, Scheiejeva bolezen pa na koncu klinične patogeneze. Med tema dvema oblikama napredovanja obstajajo prehodi različnih značilnosti. Najbolj presenetljiva lastnost je oslabljena razgradnja ogljikovih hidratov, ki se kopičijo v lizosomih celic.
Bolniki z oboleli za lovcem lahko občutijo kratko rast, povečano vranico in jetra, velike lastnosti, odebeljeno kožo, razširjen jezik in težave z dihanjem. Poleg tega se okostje pogosto spreminja na območju medenice, hrbtenice, ročnih kosti in lobanje. Možne so popkovne in [[dimeljske kile].
Zapleti
V večini primerov se simptomi ali zapleti pojavijo pri tej bolezni zelo pozno. Zaradi tega se diagnosticira pozno, zaradi česar je zgodnje zdravljenje v večini primerov nemogoče. Brez zdravljenja se med napredovanjem bolezni pojavijo različne pritožbe in poškodbe notranjih organov.
Še posebej so prizadete ledvice, jetra in vranica. Na to bolezen lahko vpliva tudi srce, kar lahko v najslabšem primeru privede do srčne smrti. Poleg tega se pojavijo poškodbe ledvic in prizadeti pogosto trpijo zaradi bolečin v prstih ali prstih. Do paralize lahko pride tudi, če so možgani s to boleznijo poškodovani. Jetra in vranica se lahko povečajo in povzročijo tudi močne bolečine.
Ni redkost, da so kosti prizadete osebe krhke in tudi boleče. Zdravljenje te bolezni se izkaže za težko. V mnogih primerih se življenjska doba prizadete osebe znatno zmanjša. Pri uporabi zdravil običajno ni posebnih zapletov. Vendar pa pozitiven potek bolezni ni mogoče zagotoviti v vsakem primeru.
Tu lahko najdete svoja zdravila
➔ Zdravila proti bolečinamKdaj morate iti k zdravniku?
Izpadanje las, težave s sklepi in motnjami organov so možni znaki bolezni lizosomalnega skladiščenja. Obisk pri zdravniku je priporočljiv, če se simptomi ponavljajo ali se pojavijo nenadoma, ne da bi ugotovili vzrok. Če so simptomi povezani s predhodno diagnosticirano encimsko okvaro ali drugo resno boleznijo, se je treba posvetovati z odgovornim zdravnikom. Nezdravljena bolezen skladiščenja lahko privede do demence, neplodnosti, nevropatij in drugih zapletov, od katerih so nekateri smrtno nevarni. Zato je treba pregledati vse možne simptome, tudi če ni posebnega suma.
Simptomi bolezni shranjevanja lizosoma se lahko pojavijo v fazah ali zahrbtno razvijejo, vendar vedno zahtevajo pregled in zdravljenje. Prizadeti ljudje je najbolje, da se direktno pogovorijo s svojim družinskim zdravnikom ali internistom. Dejansko zdravljenje poteka običajno v specialistični ambulanti za notranje bolezni, čeprav se fizioterapija ali psihoterapija lahko povežeta glede na simptome. Zlasti so terapevtski ukrepi indicirani zaradi pogosto negativnega poteka bolezni.
Terapija in zdravljenje
Glede na to, kako zgodaj je postavljena ustrezna diagnoza, je mogoče te dedne bolezni zdraviti zelo dobro z nadomestno encimsko terapijo, tako da imajo prizadeti ljudje veliko manj pritožb in s tem boljšo kakovost življenja. To nadomestno zdravljenje se uporablja v skladu s klinično sliko.
Ljudje, ki trpijo za Gaucherjevo boleznijo, nimajo "encima ß-glukocerebrosidaze", ki se proizvede biotehnološko in vstopi v bolnikovo telo. Lizosomi delujejo učinkovito in lahko absorbirajo snovi iz neposrednega okolja. Zaradi tega so umetno uporabljeni encimi spremenjeni tako, da jih je mogoče na idealen način oskrbeti z lizosomi.
Makrofagi (fagociti) razgrajujejo glukocerebrozide, ki so se nabrali v celicah. To terapijo lahko primerjamo z inzulinsko terapijo za diabetes mellitus, s to razliko, da ne manjka hormon, ampak encim, ki ni na voljo. Telo redno razgrajuje vse snovi, vključno s priloženim umetnim encimom.
Zaradi redne razgradnje snovi morajo bolniki to infuzijsko zdravljenje redno do konca življenja. Encimsko nadomestno zdravljenje ne deluje simptomatsko, ampak se neposredno bori proti vzroku dedne bolezni. Zdravniki to terapijo imenujejo vzročno. Načela terapije je treba uporabiti za vse štiri zgoraj omenjene pogoste bolezni.
Pompeške bolnike zdravijo tudi z infuzijsko terapijo. Pri tej bolezni se oskrbuje neobstoječi encim "kislina alfa glukozidaza" in pomaga razgraditi glikogen, ki se je nabral v lizosomih mišic. Pri bolnikih z vrsto bolezni "mukopolisaharidoza tipa I" lizosomalni encim "alfa-iduronidaza" ni ali ni v zadostnih količinah. Je ena redkih bolezni shranjevanja, pri kateri se molekule sladkorja kopičijo v organih in tkivih.
Če je postopek normalen, encim razgradi mukopolisaharide. Molekule sladkorja so dolgo verižne in sodelujejo pri razvoju podpornega in vezivnega tkiva, na primer kosti, kože, sklepnih tekočin in hrustanca. Če je normalen potek razgradnje moten zaradi pomanjkanja encima, se v posameznih celicah kopičijo patološki glikozaminoglikani (GAG). Prihodnje možnosti terapije so usmerjene v jemanje tablet.
Napovedi in napoved
Napoved za bolezen pri skladiščenju je slaba. Ugotovljeno je bilo, da je genetska dispozicija vzrok za zdravstveno motnjo. Zakonske zahteve zdravnikom in znanstvenikom prepovedujejo spreminjanje človeške genetike. Zaradi tega bolezen ostane vse življenje in nima možnosti za ozdravitev.
Zdravnik se osredotoča na zdravljenje simptomov, ki se pojavijo. Če se ne zdravi, se bodo sčasoma povečale različne pritožbe. Kostni sistem je poškodovan in nastanejo težave organov. V najslabšem primeru bodo notranji organi okvarili in na koncu njihova funkcija ne bo uspela. To ogroža zadevno osebo zaradi prezgodnje smrti.
Izziv bolezni je v diagnozi. Pri velikem številu bolnikov se opazne in močno zaznavne pritožbe pojavijo šele kasneje v življenju. Posledica tega je, da genetska motnja dlje časa ostane neopažena in zgodnje zdravljenje bolezni je težko. Pozneje ko je postavljena diagnoza, bolj neugoden je nadaljnji potek. V napredni fazi bolezni so notranji organi ali sklepi že močno poškodovani. Potrebni so kirurški posegi in če bolezen napreduje neugodno, lahko samo prizadeti organ reši življenje prizadeti osebi. Zgodnje zdravljenje je zato bistveno za izboljšanje prognoze.
preprečevanje
Ker gre za prirojeno genetsko okvaro, ki preprečuje izražanje encima, te bolezni ni mogoče zdraviti preventivno. Vendar bi najnovejši dosežki genskega inženiringa lahko zagotovili pristop na tem področju.
Porodna oskrba
S to boleznijo ljudje trpijo za številnimi različnimi zapleti in težavami. To praviloma zelo negativno vpliva na kakovost življenja prizadete osebe, zato je treba diagnozo postaviti že zelo zgodaj. Prej ko se posvetujemo z zdravnikom, boljši je nadaljnji potek te bolezni.
Resnost te bolezni se lahko zelo razlikuje, tako da splošna napoved pogosto ni mogoča. Prizadeti trpijo hude poškodbe notranjih organov. Prizadenejo predvsem ledvice in srce, tako da lahko otrok v prvih dneh umre, če simptomov ne odpravimo pravočasno. Na različnih delih telesa so tudi maščobe.
Še posebej so prizadeti prsti in prsti, kar lahko privede do bistveno zmanjšane estetike prizadete osebe. Praviloma se v nadaljnjem poteku pojavijo poškodbe ledvic in možganov, tako da prizadeta oseba zaradi te škode umre. Starši in sorodniki zaradi bolezni pogosto trpijo tudi za depresijo ali drugimi duševnimi motnjami.
To lahko storite sami
Bolezni lizosomalnega skladiščenja zelo pogosto potrebujejo intenzivno medicinsko oskrbo. Pogosto za samopomoč ni dovolj priložnosti. Starši prizadetih otrok pogosto doživljajo hud stres v domačem okolju, ker njihov otrok potrebuje stalno skrb in pozornost.
Klinične slike posameznih bolezni shranjevanja so različne. Obstajajo tako enostavne kot zelo težke oblike. En primer je Gaucherjeva bolezen. Pomoč staršev je pogosto omejena na hranjenje hudo invalidnega otroka. V blažjih primerih je lahko življenjska doba skoraj normalna. Kljub temu je za preprečevanje možnih zapletov potreben stalen zdravniški nadzor. Redna telesna aktivnost je ena od spremljajočih terapij, ki jo lahko izvajamo tudi doma. Poleg tega je treba urediti temeljit presejalni pregled raka. To zahteva stalen obisk zdravnika s svojim otrokom od staršev. Enako velja za druge bolezni shranjevanja lizosomov.
Pri nekaterih boleznih se poleg telesnih okvar lahko pojavijo tudi duševne okvare, ki še vedno potrebujejo posebno podporo. Pri blažjih oblikah nekaterih bolezni, kot je Hunterjeva bolezen, se sprva pojavijo le skeletne spremembe in obrazni dismorfizem. Tu pa je prizadeti bolnik pogosto sposoben voditi samostojno življenje. Vendar so tukaj potrebni tudi stalni zdravniški pregledi, da se izključijo morebitni zapleti, kot so srčno popuščanje ali bolezni dihal. Pacient se lahko s psihološkim svetovanjem spoprijema s psihološkim stresom, ki ga povzročajo fizične deformacije.


.jpg)



.jpg)



.jpg)