The Allan-Herndon-Dudley sindrom je mutacija gena SLC16A2, ki spremeni transporter ščitničnega hormona MCT8 in povzroči moten vnos jodtironina v mišično tkivo in v centralni živčni sistem. Zaradi mutacije prizadeti trpijo zaradi mišične oslabelosti, pa tudi z zamudo mobilnega in duševnega razvoja. AHDS je neozdravljiv, do zdaj so ga zdravili le z dajanjem trijodtiroacetata.
Kaj je Allan - Herndon - Dudleyjev sindrom?

Zamude pri telesnem, duševnem ali čustvenem razvoju mladostnikov in otrok so povzete kot razvojne zamude ali razvojne zaostalosti. Zamude pri razvoju so lahko različne. Na primer, sprožilec za upočasnjen razvoj lahko leži v centralnem živčnem sistemu.
Tako je na primer s sindromom Allan - Herndon - Dudley (AHDS). Poleg hude duševne zaostalosti so za sindrom značilne motnje motoričnega razvoja. Prvi opis sindroma sega v leto 1944. Klinična slika, ki jo opisujete, je dedna bolezen, to je genetska motnja.
Bolezen prizadene moške dojenčke v večini primerov, dokumentiranih do danes. Motnje v razvoju in njihove posledice so skoraj v vseh primerih jasno vidne od rojstva. AHDS je izjemno redka bolezen. Zaradi tega je bilo stanje raziskav sindroma Allan - Herndon - Dudley do zdaj precej slabo.
vzroki
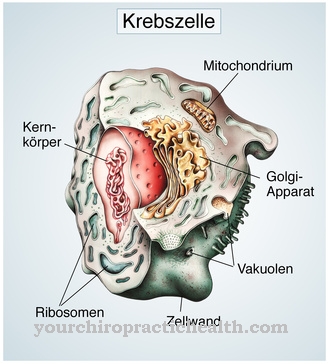
AHDS je dedna genetska motnja, ki jo povzroči mutacija gena SLC16A2. To je kodirski gen za tako imenovani transporter ščitničnih hormonov MCT8. Ta transporter posreduje vnos jodtironinov v mišično in živčno tkivo.
Zaradi mutacije pride do motenj, ko se ščitnični hormoni absorbirajo, ki uravnotežijo centralni živčni sistem in tako poslabšajo razvoj celic živčnega sistema. Mišično tkivo in možgani osiromašijo zaradi mutacije, povezane z motnjami regulacije aktivnega ščitničnega hormona, od katerega so dejansko odvisni.
Sindrom se prenaša kot dedno dedovanje, povezano z X. Ženske lahko bolezen podedujejo, vendar zaradi svoje dvojne X kromosomske strukture redko zbolijo. Bolni moški se ne morejo razmnoževati.
Čeprav je mutacija genetska, poleg tega notranjega dejavnika verjetno pri nastanku bolezni igrajo tudi zunanji dejavniki. Zaradi redkosti in omejene baze raziskav vloga teh zunanjih dejavnikov še ni dokončno razjasnjena.
Tu lahko najdete svoja zdravila
➔ Zdravila za mišično oslabelostSimptomi, težave in znaki
Allan-Herndon-Dudley sindrom je prirojena bolezen, ki se ponavadi manifestira pri malčkih ali dojenčkih. Prizadeti trpijo zaradi bolj ali manj hude mišične oslabelosti. Otroško mišično tkivo je opazno nerazvito. Šibkost mišic kmalu spremljajo deformacije sklepov.
Kontrakture so tudi pogosti spremljajoči simptomi. Mobilnost otrok čedalje bolj poslabša kontrakture in deformacije. Zaradi tega so prizadeti pogosto videti nenaravno statični ali celo negibni. Zaradi nezadostne oskrbe ščitničnih hormonov, povezanih z mutacijo, prizadeti pogosto trpijo tudi zaradi mišičnih krčev ali nehote premikajo z rokami in nogami.
Pogosto se prizadeti ne morejo samostojno gibati. V večini primerov so motorične motnje povezane s hudimi duševnimi motnjami. Na primer, večina bolnikov ne more govoriti. V posameznih primerih lahko AHDS zaznamujejo številni drugi simptomi na področju duševnega in telesnega razvoja.
Diagnoza in potek
Zdravnik ponavadi najprej sumi na AHDS v anamnezi. V laboratorijskih testih povečana raven T3 z normalnimi ravnmi FT4 in TSH kaže na sindrom Allan - Herndon - Dudley. Slikovni prikaz osrednjega živčnega sistema je običajno del diagnoze.
Mišične oslabelosti zaradi bolezni motoričnih nevronov lahko izključimo iz diferencialne diagnoze. Prognoza za bolnike z Allan-Herndon-Dudley sindromom je razmeroma slaba. Do zdaj je bolezen neozdravljiva. Študije kažejo, da lahko čas diagnoze igra ključno vlogo pri prognozi bolnika.
Zapleti
Kot vseh kromosonalno dednih motenj, tudi Allan - Herndon-Dudley sindroma ni mogoče zdraviti kurativno. Najpogostejši neželeni učinek Allana - Herndon - Dudleyjev sindrom - izrazita mišična oslabelost - se lahko zdravi s fizioterapijo. Takšno zdravljenje, namenjeno krepitvi mišic, je za bolnika lahko boleče.
Zlasti majhni otroci pogosto zavrnejo terapijo zaradi bolečin. Kljub intenzivnim treningom fizioterapija ne vodi vedno do želenega uspeha. Podobno je z govorno terapijo bolnika Allan - Herndon - Dudley. Čeprav je zmanjšanje jezikovnih sposobnosti mogoče izboljšati z intenzivnim treningom, zdravljenje ni vedno uspešno zaradi večinoma močnih duševnih okvar prizadetih.
Razočaranje s strani pacienta in velika obremenitev celotne družine sta eden najresnejših zapletov pri zdravljenju sindroma Allan - Herndon - Dudley. Mišične krče in premike okončin, na katere ne moremo vplivati, je mogoče zdraviti z dajanjem mišičnih relaksantov. Zapleti se kažejo v včasih hudih stranskih učinkih zdravil.
Poleg stresa na prebavilih je treba omeniti utrujenost, splošen občutek izčrpanosti in slabo počutje. Dolgotrajna uporaba relanksantov škoduje tudi jetrom in ledvicam. Če se sindrom Allan - Herndon - Dudley ne zdravi, prizadeti ne bodo mogli bistveno napredovati v smislu svojih duševnih ali motoričnih sposobnosti.
Kdaj morate iti k zdravniku?
V mnogih primerih neposredno zdravljenje sindroma Allan-Herndon-Dudley ni mogoče. Zaradi tega je zdravljenje večinoma simptomatsko in je namenjeno posameznim pritožbam in zamudam. Praviloma naj starši potem poiščejo zdravnika, če ima otrok mišično šibkost.
To lahko postane opazno zaradi izčrpanosti ali trajne utrujenosti. Zdravniško svetovanje je potrebno tudi, če sindrom Allan-Herndon-Dudley upočasni duševni in motorični razvoj.
Če zdravljenje ne poteka v otroštvu, lahko v odrasli dobi povzroči občutno nelagodje in omejitve. Posvetovati se je treba z zdravnikom, zlasti če bolnik ne more več govoriti. Zdravljenje je potrebno tudi pri mišičnih krčih. Če gre za akutno silo, lahko greste direktno v bolnišnico ali pokličete rešilca.
V večini primerov bo sindrom Allan-Herndon-Dudley zdravil splošni zdravnik ali pediater. Vendar pa mora posamezne pritožbe pregledati in obravnavati ustrezni specialist ali terapevt.
Zdravniki in terapevti v vaši bližini
Zdravljenje in terapija
AHDS je vzročno nezdravljiva bolezen. Ker ni na voljo terapij za odpravo primarnega vzroka, bolezen doslej ni bila ozdravljiva. Medtem napredek na področju genske terapije nakazuje, da bodo pristopi genske terapije kmalu odobreni za vsakodnevno klinično prakso.
V kolikšni meri bi bolniki s sindromom imeli koristi od odobritve, še ni razjasnjeno. Trenutno ne obstaja nobena uveljavljena ali standardizirana možnost zdravljenja za bolnike z AHDS na področju simptomatske terapije. Pred nekaj leti so raziskovalci menili, da je uporaba TRIAC možna simptomatska terapija.
TRIAC je neklasičen ščitnični hormon, trijodtiroacetat. Dajanje hormona je bilo izvedeno v klinični študiji na prizadetih otrocih, vendar ni bilo mogoče doseči vidnih rezultatov. Rezultati študije niso nujno pomembni, ker se je dajanje hormona začelo razmeroma pozno.
Zaradi tega je v letu 2014 TRIAC še vedno veljal za najboljšo možno terapijo. V enem primeru leta 2014 so med zdravljenjem z TRIAC zabeležili pomembno izboljšanje motoričnega in duševnega razvoja. Terapijo so začeli pri prizadeti osebi že v zgodnji povojih.
Rezultati dosedanjih študij kažejo, da čas začetka terapije vpliva na rezultate terapije, ki jih pri bolnikih z AHDS ne gre podcenjevati. Spremljevalne podporne terapije, kot so delovna terapija in fizioterapija ali zgodnja intervencija, se lahko teoretično uporabljajo za izboljšanje kakovosti življenja in spretnosti pacientov. Vendar pa skoraj ni dokazov o učinkovitosti takega pristopa pri bolnikih z AHDS.
Napovedi in napoved
Allan-Herndon-Dudley sindrom pri večini bolnikov povzroči številne različne simptome. V prvi vrsti prizadeti trpijo zaradi močne mišične oslabelosti. To pomeni, da običajnih dejavnosti ali športa za zadevno osebo ni več mogoče enostavno izvajati. Velike zamude pri intelektualnem in mobilnem razvoju. Bolnikova koncentracija je očitno omejena in zmanjšana.
V mišicah so še vedno močni krči in s tem pogosto neprostovoljni gibi ali trzanje. Ko napreduje sindrom Allan - Herndon - Dudley, prizadeti ne morejo več govoriti. Bolnikovo vsakdanje življenje sindrom znatno omeji in kakovost življenja se zmanjša. V nekaterih primerih so bolniki v vsakdanjem življenju odvisni od pomoči drugih ljudi.
Običajno ni mogoče zdraviti sindroma Allan - Herndon - Dudley. Zaradi tega je zdravljenje le simptomatsko. Prizadeti so odvisni od različnih terapij, ki pa ne vodijo vedno v pozitiven potek bolezni. V nekaterih primerih sindrom Allan - Herndon - Dudley omejuje življenjsko dobo prizadetih.
Tu lahko najdete svoja zdravila
➔ Zdravila za mišično oslabelostpreprečevanje
AHDS je mogoče preprečiti le z genetskim svetovanjem. Nositelji mutacije se lahko na primer odločijo, da ne bodo imeli lastnih otrok.
Porodna oskrba
Potreba po nadaljnji oskrbi gensko povzročenega sindroma Allan - Herndon - Dudley vpliva samo na moške dojenčke. Težava je v tem, da za to dedno stanje ni primerne oblike terapije. Hude posledice okvar prenosnikov ščitničnih hormonov je težko izboljšati. Poskusi olajšati prizadetim otrokom tako, da jim dajejo posebne ščitnične hormone, niso uspeli.
Težava je v tem, da je osnova bolezni običajno že vzpostavljena v materinem telesu. Trajno poškodujejo nerojenega otroka. S tega vidika se zdravljenje začne prepozno, in sicer šele po porodu. V naknadni oskrbi je mogoče zdraviti samo škodo, ki je že prisotna. Vendar upanje obstaja. Leta 2014 je bil znan primer, v katerem je dojenček z Allan-Herndon-Dudley sindromom uspešno zdravil TRIAC. Nadaljnja oskrba je bila še vedno potrebna, ker otroka ni bilo mogoče ozdraviti. Vsaj njegovi simptomi so bili ublaženi.
Allan-Herndon-Dudleyjev sindrom deloma povzroča okvarjena krvno-možganska pregrada. To kažejo študije v bolnišnici Cedars Sinai. Ščitnični hormoni zaradi pomanjkljive krvno-možganske pregrade ne morejo delovati. To velja tudi za ščitnične hormone, ki jih dajemo po rojstvu otroka. Biotehnologija ali genetske raziskave bi lahko pomagale. Vsi poskusi zdravljenja trenutno niso uspešni. To vpliva tudi na spremljanje hudo poškodovanih otrok.
To lahko storite sami
Allan-Herndon-Dudley sindrom je resno stanje, ki ga še niso učinkovito zdravili. Vendar lahko starši še vedno nekaj ukrepajo v podporo terapiji.
Najprej sta pomembna redna kognitivna vadba in telesna aktivnost. Celovita terapija, ki je, odvisno od resnosti sindroma, lahko sestavljena iz treninga govora in branja, lahko pa tudi iz splošnih možganskih vaj, ponuja tudi fizioterapevtske vaje. Trening mora biti individualno prilagojen simptomom. Starši prizadetih otrok bi morali zato zagotoviti, da so ukrepi optimalno izbrani in da otrok ne bo preobremenjen.
V primeru hudih duševnih motenj bo otrok morda potreboval stalno podporo v vsakdanjem življenju. Storitev ambulantne oskrbe je lahko za starše pomembno olajšanje. Enako pomembno je tudi bolnišnično zdravljenje, ki ga lahko podpremo doma z rednim spremljanjem simptomov.
Starši bi morali izkoristiti tudi psihološko svetovanje in po potrebi obiskati tudi skupino za samopomoč, saj stik z drugimi prizadetimi ljudmi olajša spopadanje z boleznijo. Poleg tega starši pogosto dobijo pomembne nasvete, kako ravnati z bolnim otrokom.
.jpg)





.jpg)



.jpg)