Kot Rothmund-Thomson sindrom se imenuje genetska bolezen kože. Deduje se avtosomno recesivno.
Kaj je Rothmund-Thomson sindrom?

© logo3in1 - stock.adobe.com
The Rothmund-Thomson sindrom (RTS) je ena izmed genetskih kožnih bolezni. Zlasti na področju obraza se pojavi izrazita poikiloderma, ki je povezana z ortopedskimi in oftalmološkimi očitki. Obstaja tudi večje tveganje za razvoj nekaterih vrst raka.
Ime sindroma Rothmund-Thomson sega v opis dveh zdravnikov. Prvič se je to zgodil leta 1836 nemški oftalmolog August von Rothmund (1830-1906). Leta 1936 je angleški zdravnik Matthew Sydney Thomson (1894-1969) objavil dva prispevka o dedni poikilodermi (Poikiloderma congenitale).
Ta bolezen je enaka boleznim, ki jih je opisal August von Rothemund. Zaradi tega je bil kasneje imenovan Rothmund-Thomson sindrom. Rothmund-Thomson sindrom lahko uvrstimo med redke bolezni. Do leta 2014 je bilo registriranih le 300 primerov bolezni. Sindrom se pojavlja v družinah, kar je značilno za dedne bolezni.
Prizadenejo predvsem krvni sorodniki ali majhne skupnosti. Med obema spoloma obstaja ravnovesje glede na število bolezni. Zaradi majhnega števila primerov ni mogoče podati natančnih informacij. Poleg tega Rothmund-Thomson sindrom ni posebej prizadet nobene etnične skupine. Nosilna frekvenca mutacije ni znana.
vzroki
V medicini je sindrom Rothmund-Thomson razdeljen na podformi RTS-1 in RTS-2. Do zdaj še ni bilo mogoče razjasniti, kaj povzroča RTS-1. RTS-2 je mogoče zaslediti do heterorozne mutacije, ki izvira iz gena RECQL4. Prizadeti gen je kodiran za helikazo.
Najpogostejša mutacija je nesmiselna mutacija. Med 60 in 65 odstotki vseh bolnikov so okvare gena RECQL4 vzrok za razvoj sindroma Rothmund-Thomson. Obe vrsti RTS-1 in RTS-2 se podedujeta na avtosomno recesivni način.
Simptomi, težave in znaki
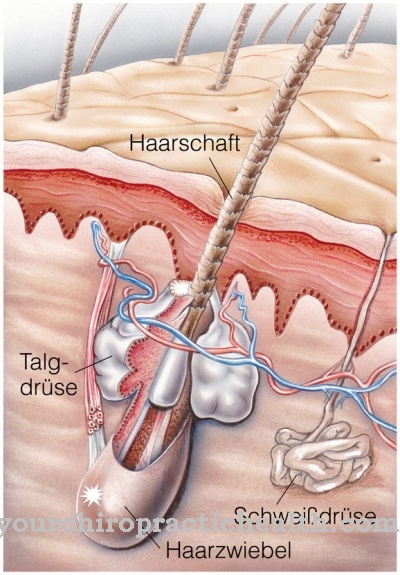
Značilna značilnost Rothmund-Thomsonsovega sindroma je nezgrešljiv izpuščaj na obrazu, ki je poikiloderma. Ta izpuščaj je glavni simptom genetske kožne bolezni, Poikiloderma pa bolezen razlikuje tudi od sindroma RAPADILINO.
Druge značilnosti sindroma Rothmund-Thomson so redki lasje na glavi, pogosto pomanjkanje trepalnic ali obrvi, kratka rast, okvare okostja, mladostniške katarakte in pojav radialne klubske roke, poleg tega pa prizadeti prezgodaj starajo in razvijejo nagnjenost k raku.
Simptomi se med RTS-1 in RTS-2 razlikujejo. Pri RTS-1 se pojavijo poikiloderma, ektodermalna displazija in juvenilna katarakta oči. Bolniki s podtipom RTS-2 trpijo tudi zaradi poikiloderme. Poleg tega obstajajo prirojene deformacije kosti in povečano tveganje za pojav osteosarkomov, malignih kostnih tumorjev v adolescenci. V nadaljnjem življenju lahko privede tudi do kožnega raka.
Malformacije okostja pri sindromu Rothmund-Thomson so med drugim opazne po sedlu, nosu, radialni klubski roki ali izrazitem čelu. V nekaterih primerih pa jih lahko zaznamo le z rentgenskimi žarki.
Diagnoza in potek bolezni
Prav tako je treba razlikovati med obema oblikama Rothmund-Thomson sindroma v diagnostiki. Za razvoj RTS-1 ni bilo mogoče najti molekularnih genetskih sprožilcev. Zaradi tega diagnoza temelji na prisotnih simptomih. V ta namen je bila izdelana posebna tabela za ocenjevanje, ki se uporablja za zbiranje različnih pritožb zaradi bolezni.
S pomočjo točkovnih vrednosti lahko preiskovani zdravnik ugotovi, ali je prisoten RTS-1. Če obstaja sum RTS-2, je mogoče določiti mutacijo v genu RECQL4 z genetskim testom. To ponavadi tudi dokazuje RTS-2. Rothmund-Thomson sindrom je treba upoštevati, če ima bolnik osteosarkom. Za RAPADILINO sindrom in Baller-Geroldov sindrom je treba postaviti diferencialno diagnozo, saj lahko pri teh boleznih najdemo tudi mutacije v genu RECQL4. Sindrom Rothmund-Thomson poteka drugače.
Kljub vidnemu procesu prezgodnjega staranja imajo pacienti relativno normalno življenjsko dobo, pod pogojem, da ni malignega raka. Pri nekaterih bolnikih obstaja večje tveganje za razvoj sekundarnih malignomov. V teh primerih je prognoza odvisna od kakovosti presejalnih pregledov in zdravljenja raka. Če se pojavijo osteosarkomi, je 5-letna življenjska doba bolnikov med 60 in 70 odstotki.
Zapleti
Pri sindromu Rothmund-Thomson ljudje trpijo zaradi številnih različnih kožnih stanj. Ti očitki zelo negativno vplivajo na estetiko, tako da se mnogi prizadeti počutijo neprijetno in trpijo zaradi kompleksov manjvrednosti ali zmanjšane samozavesti. Posledica tega je lahko tudi depresija ali druge psihološke pritožbe. Rothmund-Thomson sindrom lahko privede do ustrahovanja ali draženja, zlasti pri otrocih.
Poleg tega je na okostju tudi kratek stas in različne nepravilnosti. Bolniki trpijo zaradi omejene gibljivosti in v nekaterih primerih zaradi razvoja kostnih tumorjev. Kožni rak se lahko razvije tudi kot posledica bolezni in lahko znatno skrajša življenjsko dobo prizadete osebe. Poleg tega so v mnogih primerih starši ali sorodniki prizadeti tudi zaradi simptomov Rothmund-Thomson sindroma in trpijo tudi zaradi psiholoških pritožb.
Ker vzročnega zdravljenja za sindrom ni, lahko izvajamo le simptomatsko zdravljenje. Zapletov ni. Vendar so prizadeti odvisni od presaditve matičnih celic, da bi premagali raka. Popolnoma pozitiven potek bolezni praviloma ni mogoče doseči.
Kdaj morate iti k zdravniku?
Ker je Rothmund-Thomson sindrom dedna kožna bolezen z družinsko razširjenostjo, so stiki z zdravnikom na začetku življenja neizogibni. Vendar poikiloderma v obraznem predelu, povezana z boleznijo, ni edini simptom, ki oteženim življenjem otežuje življenje.
Ljudje z Rothmund-Thomson sindromom trpijo tudi zaradi ortopedskih ali oftalmoloških zapletov. Ljudje obeh spolov imajo tudi večjo verjetnost, da bodo zboleli za rakom. Vendar pa se podtipi Rothmund-Thomson sindroma pojavljajo razmeroma redko. Na svetu ni dokumentiranih več kot 300-320 primerov.
Glede na redkost Rothmund-Thomssonovega sindroma je razmeroma težko najti zdravnika specialista, ki bi mu lahko postavil diagnozo Rothmund-Thomson sindroma. Potrebna je diferenciacija od drugih sindromov s podobnimi simptomi. Zdravstveni posegi so v večini primerov nepogrešljivi, tudi če ni možnosti za ozdravitev. Rothmund-Thomson sindrom lahko zdravimo le simptomatsko. Za tesno sodelovanje si bodo prizadevali strokovnjaki in specialisti iz različnih strok.
Sodelovanje dermatologov, ortopedov, oftalmologov, kirurgov ali onkologov je najboljši način za pomoč prizadetim pri doseganju boljše kakovosti življenja.
Terapija in zdravljenje
Trenutno ni terapije za vzrok Rothmund-Thomson sindroma. Zaradi tega je zdravljenje omejeno na lajšanje simptomov. Ker so simptomi izredno zapleteni, je priporočljivo, da bolnika zdravimo interdisciplinarno. To pomeni, da terapijo izvajajo zdravniki različnih specialnosti. V primeru sindroma Rothmund-Thomson to vključuje kirurge, ortopede, oftalmologe, onkologe in dermatologe (dermatologe).
Simptomatska terapija vključuje letni oftalmološki pregled, zdravljenje telangieektazije in radiološke preiskave za nadzor nad osteosarkomi. Presaditev matičnih celic je možna nova možnost terapije, vendar je še vedno v kliničnih preskušanjih in je bila doslej opravljena le na dveh bolnikih. En bolnik je opravil alogensko presaditev kostnega mozga. V drugem primeru je bilo zdravljenje izvedeno z uporabo matičnih celic popkovnične krvi.
Tu lahko najdete svoja zdravila
➔ Zdravila za pordelost kože in ekcemepreprečevanje
Žal ni mogoče sprejeti preventivnih ukrepov proti izbruhu Rothmund-Thomson sindroma. Stanje je ena od dednih bolezni, katere vzrok še vedno ni znan.
Porodna oskrba
Nadaljnja oskrba za Rothmund-Thomson sindrom je odvisna od podtipa bolezni in njenega poteka, pa tudi od izbranih ukrepov zdravljenja in pacientove ustave. Na splošno bodo nadaljnji ukrepi pregledali različne simptome in razpravljali o naslednjih korakih. Kirurški poseg pogosto predstavlja veliko breme za pacienta.
Med spremljanjem se pregledajo kirurške brazgotine, da se ugotovi uspešnost zdravljenja. Poleg tega paciente spodbujamo k počitku. Za dolgotrajne bolezni bo morda potrebno zdravljenje s protibolečinskimi zdravili. Poleg tega pacienti včasih potrebujejo psihološko podporo. Del nadaljnje oskrbe je tudi izobraževanje o napredku novih metod terapije, kot sta presaditev matičnih celic ali presaditev kostnega mozga, ki so jih do zdaj uspešno izvajali pri nekaterih bolnikih.
Nadaljnje spremljanje vključuje rutinski pregled različnih telesnih funkcij, ki jih je prizadel rak. Če ne pride do zapletov, lahko bolnika nato odpustimo. Ker gre za izredno redko stanje, ki ga je mogoče zdraviti le simptomatsko, je za bolnika priporočljivo natančno zdravstveno spremljanje tudi po okrevanju. Nadaljnjo nego Rothmund-Thomson sindroma izvaja dermatolog, ortoped, oftalmolog, onkolog ali kirurg, odvisno od simptomov.
To lahko storite sami
Ker je Rothmund-Thomson sindrom povezan s povečanim tveganjem za raka, mora prizadeta oseba v rednih presledkih neodvisno pregledati svoje telo glede nepravilnosti ali nepravilnosti na koži. Poleg zdravstvenih obiskov za zdravniške preglede je priporočljivo poiskati pomoč drugih ljudi v vsakdanjem življenju, da bi razjasnili področja, ki so težko dostopna za morebitne spremembe videza kože.
Ker genetska bolezen že v prvih letih življenja kaže nepravilnosti, je treba odraščajočega otroka v zgodnji fazi obvestiti o obstoječi bolezni in njenem nadaljnjem poteku. Pri vsakodnevnem spopadanju z motnjo je pomembno, da se izognemo nenadnim situacijam in neprijetnim presenečenjem. Zato je treba tesno socialno okolje obvestiti tudi o obstoječih okvarah zdravja. Ker obstajajo vizualne spremembe, je pomembno okrepiti samozavest v otrokovem razvoju in rasti.
Ker se za izboljšanje stanja pogosto uporabljajo kirurški posegi, je duševna podpora otroku prav tako pomembna kot krepitev imunskega sistema. Organizem potrebuje obrambo, da se spopade z operacijo. Duševno moč je mogoče graditi v sodelovanju s psihoterapevtom. Poleg tega ponujene terapije pomagajo, da lahko čustveno predelamo izzive bolezni. V nekaterih primerih morajo starši razmisliti tudi o psihoterapevtskem zdravljenju.

.jpg)
.jpg)
.jpg)




.jpg)