The Crouzonov sindrom, tudi Crouzonova bolezen imenovan, je ena izmed več znanih, gensko povzročenih kraniosinostoz, pri katerih se lobanjski šivi prezgodaj okostelijo, tako da je rast lobanje motena in se lahko pojavijo značilne nepravilnosti in adhezije na glavi in obrazu. Mentalni razvoj ljudi, ki jih prizadene Crouzonov sindrom, je običajno normalen.
Kaj je Crouzonov sindrom?

© royaltystockphoto - stock.adobe.com
Tudi Crouzonov sindrom Kraniofacialna dizostoza Crouzon imenovan, je ena izmed več znanih kraniosinostoz. Za bolezen je značilna zgodnja okostitev lobanjskih šivov, od katerih se nekateri začnejo prenatalno. Kostitev pomeni, da se možgani v fazi rasti ne morejo zlahka širiti pod lobanjsko kapico, ki običajno "raste tudi z otrokom". Namesto tega lobanja raste predvsem na še ne okostenelih lobanjskih šivih, tako da, če jih ne zdravimo, pride do značilnih nepravilnosti.
Pri Crouzonovem sindromu se koronarni, alfa in sagittalni šivi sprva okostenijo. Brez zdravljenja ali korektivnih kirurških posegov se pojavijo značilne nepravilnosti lobanje stolpnic in obraza, kot sta pretirano olajšanje oči (hipertelorizem) in štrleče oči (eksoftalmos). Poleg neskladnosti zob je treba pri Crouzonovem sindromu pričakovati gluhost, ker ima zunanji slušni kanal zamašitev, atrezija slušnega kanala in / ali kostnice niso v celoti razvite, tako da pride do okvare sluha.
vzroki
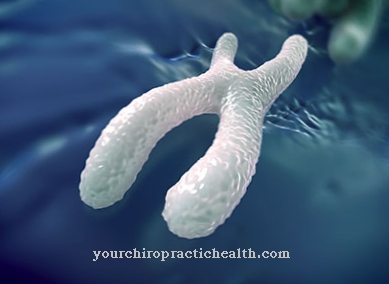
Crouzonov sindrom povzroča izključno mutacija v lokusu gena 10q26 na kromosomu 10. Na kromosomu 10 je približno 1.200 genov, ki vsebujejo 4% do 4,5% človeške celične DNK. Gen 10q26 je odgovoren za kodiranje "receptorja rastnega faktorja 2 fibroblasta" (FGFR2). Učinki te specifične mutacije genov so v opazovanem območju različno izraziti.
Mutacija genov je podedovana kot avtosomno prevladujoča lastnost. To pomeni, da Crouzonov sindrom ni specifičen za spol, zato lahko enako prizadene moške in ženske, kar pomeni, da se bo bolezen zagotovo pojavila, tudi če na genetsko okvaro na mestu 10q26 vpliva samo en starš. Najbolj opazen učinek te genetske okvare je prezgodnja okostitev lobanjskih šivov. Lobanjski šivi predstavljajo rastne plošče kostnih plošč čelne kosti (Os frontale), parietalne kosti (Os parietale) in okcipitalne kosti (Os occipitale).
Če se šivi v fazi rasti okostenijo, se lobanja ne more enakomerno razširiti in možgani povzročajo naraščajoči rastni pritisk, kar vodi do značilnih deformacij lobanje.
Simptomi, težave in znaki
Poleg že opisanih simptomov Crouzonovega sindroma, kot so lobanja stolpa, štrleče oči in široko olajšanje oči, obstajajo tudi drugi znaki, ki kažejo na prisotnost Crouzonovega sindroma. To so štrleči okosteni šivi lobanje, počepi v očeh in strabizem. Strabizem je pomanjkanje koordinacije očesnih mišic.
Oči ne morejo biti vzporedne ali poravnane na skupnem predmetu. Hipoplazija zgornje čeljusti in štrleča spodnja ustnica sta tudi stranska učinka Crouzonovega sindroma. Simptomatska maksilarna hipoplazija, znana tudi kot maksilarna retrognacija, je brada, ki štrli daleč od maksile.
Na splošno je rezultat podoba konkavnega obraza. Praviloma simptomatski manifestacije Crouzonovega sindroma niso omejene na lobanjo, pojavijo pa se tudi drugi "povezani" problemi. Treba je omeniti humero-radialno sinostozo, delno okostjevanje v ramenskem sklepu in subluksacijo v komolčnem sklepu.
Diagnoza in potek
Sum na možno prisotnost Crouzonovega sindroma lahko nastane prenatalno na podlagi družinske anamneze. Zunanje vidni simptomi naredijo diagnostično slikanje skoraj odveč. Če obstaja dvom, ali je prisoten Crouzonov sindrom, lahko genetska analiza zagotovi informacije. Potek bolezni se razlikuje od osebe do osebe, zlasti v primeru pridruženih kliničnih slik.
Če bolezni ne zdravimo, se ponavadi glavni simptomi pojavijo med glavno fazo rasti lobanje in možganov. Ko se faza rasti konča, jasno vidne deformacije na glavi in obrazu ostanejo vse življenje, razen če se izvajajo kirurški posegi.
Zapleti
Praviloma je pri Crouzonovem sindromu močno prizadeto zlasti oblikovanje lobanje, kar ima za posledico okostenje lobanjskih šivov. To lahko povzroči ogromne deformacije na glavi, ki močno vplivajo na bolnikov videz. Večina ljudi trpi zaradi zmanjšane samopodobe in se počutijo neprivlačne.
Crouzonov sindrom lahko privede tudi do socialnih težav, kar še posebej velja pri otrocih in mladih. Spremenjen videz lahko privede do nasilništva. Pri Crouzonovem sindromu duševni razvoj v večini primerov ni oslabljen. Na oči vpliva tudi Crouzonov sindrom, tako da lahko pride do strabizma. To vodi v težave pri usklajevanju.
Zapleti nastanejo, ko se Crouzonov sindrom v otroštvu ne zdravi kirurško. Samo zdravljenje je možno le kot kirurški ukrep in je namenjeno predvsem popravljanju nepravilnosti. Predvsem pa se ustvarja prostor za rastoče možgane. Vendar popolno ozdravitev sindroma ni mogoče. Zmanjšanje življenjske dobe ni, dokler med operacijo ni posebnih zapletov.
Kdaj morate iti k zdravniku?
V večini primerov je Crouzonov sindrom prepoznan takoj po rojstvu ali celo pred rojstvom, tako da v večini primerov dodatna diagnoza ni potrebna. Vendar pa se je treba posvetovati z zdravnikom za zdravljenje posameznih pritožb in napak.
Zlasti, če ima otrok mehkec, se je treba za odpravo te pritožbe posvetovati z zdravnikom. Na Crouzonov sindrom lahko vplivajo tudi mišice v obrazu, zato je obisk zdravnika nujen, če bolnik ne more oblikovati neodvisnega izraza obraza.
Osifikacija sklepov lahko kaže tudi na sindrom in ga je treba pregledati. Praviloma simptome lahko pregleda in diagnosticira pediater ali splošni zdravnik. Nadaljnje zdravljenje je odvisno od resnosti simptomov, tako da bodo morda potrebni kirurški posegi.
Če ima otrok ali njihovi svojci in starši psihološke pritožbe zaradi Crouzonovega sindroma, se je treba posvetovati tudi s psihologom, da se izognemo nadaljnjim pritožbam in zapletom.
Zdravniki in terapevti v vaši bližini
Zdravljenje in terapija
Zdravljenje Crouzonovega sindroma je v glavnem sestavljeno - če sploh - iz korektivnih operacij. Znane so tri različne kirurške tehnike, ki jih nudijo specialne ambulante. Sprednje orbitalno napredovanje je sestavljeno iz razrezanja sprednje strani lobanje, vključno s čelom, v lobanji in ponovnega pritrjevanja, tako da imajo možgani prostor za potrebno rast.
Ponovno pritrditev lobanje je v osnovi mogoče s pomočjo titanovih plošč, z vpojnim sistemom plošč ali z vpojnim materialom za šivanje. Katera metoda se uporablja, je odvisno od pogojev, ki se srečujejo med operacijo. Operacije na koščeni obrazni lobanji so običajno bolj zapletene in so znane kot Le Fort III osteotomija. V nekaterih primerih lahko to popravi tudi preširok položaj oči.
Tretji postopek, distrakcijska osteogeneza, omogoča postopen premik lobanjskih plošč. Naprave za distrakcijo, ki so namenjene določenim področjem lobanje, se uporabljajo kirurško, le nekaj dni po operaciji lahko kostne plošče vsak dan odstranimo do milimetra s pomočjo vgrajenega sistema fiksacije. Kost zapolni vrzel s kalusnim tkivom, ki se kasneje okosteni, kar ustvari nekakšno umetno rast lobanje.
Napovedi in napoved
Crouzonov sindrom je treba vedno zdraviti. Če sindroma ne zdravimo, običajno vodi v smrt. Zdravljenje lahko temelji le na simptomih sindroma in ne sme biti vzročno.
Malformacije se odpravijo s pomočjo kirurških posegov. Zgodnja diagnoza in zdravljenje zelo pozitivno vplivata na nadaljnji potek bolezni. Zgodnja operacija daje možganom dovolj prostora za zdravo rast, da v bolnikovem življenju ne pride do nadaljnjih omejitev ali nelagodja.
Zadevna oseba po postopku običajno ne trpi več pritožb in ni zapletov. Tudi duševni razvoj pacienta ne moti bolezen, če se zdravi zgodaj. Tudi po uspešnem zdravljenju so priporočljivi redni pregledi, da se izognemo nadaljnjim simptomom.
Praviloma Crouzonov sindrom nima negativnega vpliva na življenjsko dobo prizadete osebe, če je odkrit zgodaj in zdravljen v celoti. Če s Crouzonovim sindromom ne zdravimo, bo to privedlo do nadaljnjih nepravilnosti v obrazu in s tem do omejitev v življenju prizadete osebe. Posebej so prizadeti ramenski sklepi in oči.
Preprečiti
Ker je pojav Crouzonovega sindroma genetski, neposrednih preventivnih ukrepov ni znanih. Ni ukrepov, ki bi lahko bolezen preprečili sami. Če obstaja sum vzročne genetske okvare, so pomembni preventivni ukrepi za zmanjšanje učinkov bolezni, zlasti v fazi rasti.
Preventivni ukrepi so sestavljeni iz rednega vizualnega pregleda lobanjske kapice in preverjanja intrakranialnega tlaka, da bi možganom ponudili možnost normalne rasti pod lobanjsko kapico z operacijo na lobanji.
Porodna oskrba
S Crouzonovim sindromom v večini primerov niso na voljo posebni nadaljnji ukrepi prizadetim. S to boleznijo je bolnik odvisen predvsem od hitre diagnoze in naknadnega zdravljenja, saj se le tako lahko izognemo nadaljnjim zapletom ali nadaljnjim poslabšanjem simptomov.
Zato se je treba ob prvih znakih bolezni posvetovati z zdravnikom. Prej se začne zdravljenje, boljši bo običajno nadaljnji potek te bolezni. Ker gre za genetsko bolezen, je treba, če želite imeti otroke, vedno najprej opraviti genetsko svetovanje. To lahko prepreči, da bi se Crouzonov sindrom ponovno pojavil pri potomcih.
Zdravljenje poteka s kirurškim posegom. Prizadeta oseba bi se po tem postopku zagotovo morala spočiti in skrbeti za svoje telo. Da se ne bi po nepotrebnem obremenjevali telesa, se vzdržite napornega napora ali stresnih in telesnih dejavnosti. Poleg tega je pogosto zelo pomembna podpora lastne družine ali prijateljev in znancev. V večini primerov ta bolezen ne zmanjša življenjske dobe prizadete osebe.
To lahko storite sami
Ko se otrok rodi s Crouzonovim sindromom, so starši prvi izziv. Pomembno je, da bolnik opravi celovito diagnozo. Izdelati je mogoče terapevtske načrte, ki vključujejo tako kirurške posege kot tudi izbiro medicinskih pripomočkov. Zlasti operacije lobanje je treba izvesti čim prej.
Vendar pa bolniki s Crouzonovim sindromom ne potrebujejo le obsežne medicinske obravnave. Običajno so strmeli, marginalizirani ali ustrahovani že od malih nog. Tu lahko pomaga empatična psihoterapija, v katero naj bi bili vključeni tudi starši in sorojenci.
Pogosto je koristno, da starši in bolniki vzpostavijo stik z drugimi prizadetimi osebami. Za to obstaja več možnosti.Na primer, spletno mesto "Sindrom pobude za starše" s strokovnim sindromom in sorodnimi malformacijami, npr. "(Www.apert-syndrom.de) ponuja tudi informacije o Crouzonovem sindromu in ponuja tudi vsakoletni trening kot družinsko srečanje.
Približno osemdeset bolnikov s Crouzonovim sindromom je trenutno registriranih na spletni strani "Diseasemaps". Če se pridružite temu - uglednemu spletnemu mestu, se lahko obrnete na posamezne paciente v ustreznih državah (www.diseasemaps.org/de/crouzon-syndrome).
Ker se sindrom podeduje na avtosomno prevladujoč način, morajo bolniki s Crouzonovim sindromom pri poskusu spočetja poiskati genetski nasvet.
.jpg)
.jpg)





.jpg)