Kot Lenz-Majewski sindrom zdravnik pozna vrsto hiperostotične kratke postave, ki je povezana s cutix laxo in osteosklerozo. Sindrom temelji na mutaciji gena PTDSS1 na genskem lokusu 8q22.1. Vzročena terapija za prizadete še ni na voljo.
Kaj je Lenz Majewski sindrom?

© deepagopi2011 - stock.adobe.com
The Lenz-Majewski sindrom je posebna in izjemno redka oblika kratke rasti. Kompleks simptomov je bil doslej opisan le v devetih primerih. Kot sindrom iz skupine hiperostotičnih kratkih stanj kompleks ne zaznamuje le kratka rast, temveč tudi značilne obrazne poteze in cutis laksa. V novo opisanih primerih so opazili tudi progresivno sklerozo kosti.
Progresivna skleroza je znana tudi kot osteoskleroza in ustreza utrjevanju kostnega tkiva. Domneva se, da je razširjenost Lenz-Majewskega sindroma le en primer na milijon ljudi. Klinični izrazi so sopomenke za simptomski kompleks Braham-Lenzov sindrom in Lenz-Majewski hiperostotični škrat.
Prvi sinonim sega do prvega deskriptorja Brahama. V svojih pripombah v 20. stoletju je sindrom še vedno označil za Camurati-Engelmannov sindrom. Malo pozneje sta nemška človeška genetičarka Lenz in pediater Majewski opravila začetno razmejitev. Ime bolezni kot Lenz-Majewski sindrom se nanaša na to prvo razmejitev proti koncu 20. stoletja.
vzroki
Lenz-Majewski sindrom ima genetski vzrok. Do zdaj opaženi primeri se ne pojavljajo sporadično, vendar so podvrženi avtosomno prevladujočemu dedovanju. Zlasti starost očetov prizadetih otrok navaja predvsem avtosomno prevladujoče nove mutacije kot glavni vzrok simptomov.
Medtem je bil kljub redkim opisanim primerom ugotovljen gen povzročitelja. Sindrom verjetno temelji na mutaciji gena PTDSS1, ki se nahaja na genskem lokusu 8q22.1. Gen kodira za protein, imenovan fosfatidilserin sintaza 1.
Zaradi mutacije gena protein izgubi svojo funkcijo, ki je sestavljena iz tvorbe fosfatidilserina. Fosfatidilserini so pomembni fosfolipidi, ki jih štejemo med sestavine membrane. Zdi se, da ta povezava sproži skrajno kratek stavek s patološko spremenjenimi telesi vretenc.
Simptomi, težave in znaki
Bolniki z Lenz-Majewskim sindromom trpijo zaradi različnih kliničnih meril. Bolezen se manifestira v zgodnji povojih in se v tem času pojavi kot nezmožnost uspevanja. Bolniki se pojavljajo v predčasni starosti in progeroidi. Vaši lobanjski šivi so nenavadno široki. Enako velja za njen fontanel. Pogosto takoj po rojstvu je opazen kraniofacialni dismorfizem.
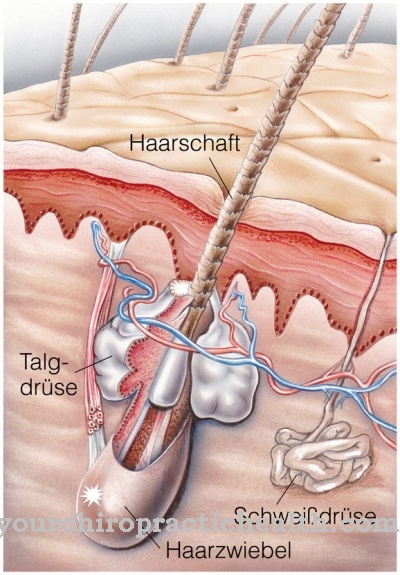
Večina bolnikov ima izrazito in izjemno široko čelo. Poleg tega pogosto obstaja hipertelorizem. Solzni kanali prizadetih so pogosto bolj ali manj ovirani pri svojem poteku. Auricles so videti pretirano velike in opazno ohlapne. Običajno obstajajo pomanjkljivosti v sklenini prizadetih, ki kasneje spodbujajo zobno gnilobo. Staran videz prizadetih lahko zasledimo do tako imenovanega cutis laxa.
Ta pojav ustreza tanki in izsušeni koži, za katero so vse bolj značilne žile in je povezana s kilami, kriptorhidizmom ali hipospadijo. Večina bolnikov trpi za duševno zaostalostjo. Na prstih se sindrom manifestira kot membranske sinandalije, ki običajno prizadenejo prostor med drugim in petim prstom.
Huda kratka rast je najbolj značilen simptom Lenz-Majewskega sindroma. V določenih okoliščinah brahidaktilija vpliva tudi na okončine prizadete osebe in se zdi, da so skrajšane.
Diagnoza in potek bolezni
Za postavitev prvotne suma diagnoze Lenz-Majewskega sindroma zdravnik običajno potrebuje le klinično sliko. Poleg tega je mogoče urediti rentgensko sliko. Slike prikazujejo značilne kriterije, kot je progresivna osteoskleroza na lobanjskih kosteh in telesih vretenc. Ta znak je lahko povezan s širokimi rebri ali ovratnicami.
Poleg tega je v določenih okoliščinah na rentgenski sliki mogoče opaziti diafizno sklerozo s širjenjem kosti. Za sindrom govori tudi hipoplazija srednjih falang. Glede diferencialne diagnoze mora zdravnik sindrom razlikovati od simptomskih kompleksov s podobnim videzom, na primer sindrom Camurati-Engelmanna, kraniodiafizno displazijo ali kraniometafizno displazijo.
Za potrditev domnevne diagnoze se lahko izvede molekularno genetski test. Če pacient resnično trpi za Lenz-Majewskim sindromom, ta analiza zagotavlja dokaze o genetski mutaciji.
Zapleti
Pri sindromu Lenza-Majewskega prizadeti predvsem trpijo zaradi kratke rasti. Pri otrocih lahko ta očitek povzroči težave z duševnim zdravjem ali ustrahovanje in draženje. S to boleznijo se znatno zmanjša kakovost življenja prizadete osebe. Poleg tega ni redko, da se pojavi zobna gniloba in druge okvare.
Brez zdravljenja prizadeti trpijo hud zobobol in drugi neprijetni simptomi v ustni votlini. Ni redko, da sindrom Lenz-Majewski privede do duševne zaostalosti, tako da so bolniki v vsakdanjem življenju odvisni od pomoči drugih ljudi. Pogosto starši in sorodniki prizadetih trpijo tudi zaradi simptomov psiholoških pritožb ali depresije.
Poleg tega se lahko posamezne okončine zaradi bolezni skrajšajo, kar lahko privede tudi do različnih omejitev v življenju. Lenz-Majewskega sindroma ni mogoče zdraviti vzročno. Zaradi tega je zdravljenje usmerjeno predvsem v zmanjšanje posameznih simptomov. Zapletov ni, potek bolezni pa ni povsem pozitiven. Praviloma so prizadeti zaradi sindroma Lenz-Majewski vse življenje odvisni od pomoči drugih ljudi.
Kdaj morate iti k zdravniku?
Bolniki z Lenz-Majewskim sindromom imajo različne simptome, ki jih je treba razjasniti. Ker je bolezen dedna, lahko simptome diagnosticiramo kmalu po rojstvu. Starši se morajo tesno posvetovati z zdravnikom, če obstajajo kakšni simptomi ali zapleti. Če otrok razvije čustvene težave kot posledice nepravilnosti in sprememb na koži, mora poklicati terapevta. Zaplete zaradi kakršnih koli duševnih pritožb mora razjasniti tudi specialist.
Za starše je najbolje, da ostanejo v stiku s specialistično ambulanto za genetske bolezni. Če je otrok pokazal znake motnje vida ali nelagodje v ustih, se je treba posvetovati z drugimi zdravniki. Karakterističen kratek stanek mora zdraviti ortopedski kirurg. Če se sindrom Lenz-Majewski zdravi zgodaj, se simptomi lahko močno ublažijo. Zato je potrebna zgodnja diagnoza, tudi če simptomi hiperostotične kratke postave na začetku morda niso zelo izraziti. Po začetnem zdravljenju otrok še vedno potrebuje pomoč fizioterapevtov, psihologov in zdravnikov.
Zdravljenje in terapija
Za bolnike z Lenz-Majewskim sindromom za zdaj ni na voljo vzročnih terapevtskih pristopov. Vendar so metode genske terapije trenutno predmet medicinskih raziskav. Glede na napredek te raziskovalne teme lahko v prihodnosti obstaja genska terapija za vzročno zdravljenje. Trenutno pa morajo biti prizadeti zadovoljni s simptomatskim zdravljenjem.
To simptomatsko zdravljenje lahko vključuje na primer kirurško odpravljanje nepravilnosti. Takšnega popravljanja ni vedno treba izvesti, ampak je omejen na sndaktilije, ki pacienta omejujejo v njegovem vsakdanjem življenju. Postopno utrjevanje kosti se lahko v nekaterih okoliščinah upočasni s prehranskimi in medicinskimi posegi.
Osteosklerozo je treba stalno in natančno spremljati, da se hitro zlomijo kateri koli zlomi. Zaradi duševnih in intelektualnih primanjkljajev prizadetim bolnikom običajno priporočamo zgodnjo intervencijo. Kot del tega zgodnjega posredovanja se lahko v vsakem primeru zmanjšajo primanjkljaji na intelektualni ravni, tako da so zmerni in komaj opazni. Starši prizadetih otrok bi morali v najboljšem primeru dobiti podrobne nasvete o možnih možnostih financiranja.
Napovedi in napoved
Prognoza za Lenz-Majewski sindrom je slaba. Glede na trenutni medicinski in znanstveni status ni terapevtske možnosti, ki bi privedla do okrevanja. Vzrok za zdravstveno motnjo temelji na genetski okvari. Ker sprememb v človeški genetiki iz pravnih razlogov ni mogoče izvesti, mutacije ni mogoče popraviti.
Nadaljnji potek bolezni je odvisen od resnosti posameznih pritožb. Kljub temu je pri bolniku s sindromom kakovost življenja na splošno precej omejena. Tudi če so simptomi blagi, obstajajo nepravilnosti v procesu rasti. Za bolezen je značilen kratek stas. Tega se ne da popraviti niti z dajanjem hormonov. Vizualna napaka vodi v stanje čustvene stiske. Zaradi tega se pri prizadetih poveča tveganje za psihološke zaplete.
Pri hudem poteku bolezni obstajajo tudi kognitivne omejitve poleg fizičnih nepravilnosti. Duševno zaostalost lahko že v prvih letih življenja podpremo z zgodnjo podporo. Skupna kognitivna uspešnost se nato izboljša, vendar obstaja možnost, da bodo omejitve v ravni uspešnosti ostale za vse življenje. Primanjkljaje je mogoče zmanjšati s pomočjo terapevta in podpore sorodnikov. To nekaterim pacientom omogoča dobro življenje.
preprečevanje
Lenz-Majewskega sindroma doslej ni mogoče preprečiti, saj simptomski kompleks povzroča nova genetska mutacija iz neznanega razloga.
Porodna oskrba
Ker je Lenz-Majewski sindrom neozdravljiv, je potrebna redna in celovita nadaljnja oskrba. Prizadeti običajno trpijo zaradi številnih zapletov in pritožb, kar lahko v najslabšem primeru privede do smrti zadevne osebe. Zato je treba bolezen prepoznati zelo hitro, da bi vsebovali nadaljnje pritožbe ali zaplete.
Bolniki bi morali redno obiskati zdravnika, ki bo pregledal nastavitve zdravil in možne neželene učinke. Ker je bolezen lahko zelo stresna za prizadeto osebo in njihove svojce, se lahko priporoči psihološka podpora, da se na splošno olajša trpljenje.
To lahko storite sami
Sindroma Lenza-Majewskega ni mogoče zdraviti s samopomočjo. Praviloma tudi neposredno medicinsko zdravljenje sindroma ni mogoče, saj gre za dedno bolezen.
Če ima zadevna oseba intelektualni primanjkljaj, jih je treba odpraviti z intenzivno podporo. Prej ko se začne to financiranje, večja je verjetnost kompenzacije teh primanjkljajev. Starši lahko z otrokom delajo tudi različne vaje v svojem domu, da bi spodbudili in okrepili duha. Otroku pa je treba nuditi tudi intenzivno podporo v vrtcu in šoli.
Utrjevanje kosti lahko olajšate z jemanjem zdravil. Pomembno je zagotoviti, da se ga redno jemlje, čeprav morajo biti starši pozorni na to pravilnost, zlasti pri otrocih. Utrjevanje se lahko ustavi tudi s prilagojeno prehrano. Tu pa je treba upoštevati natančna navodila zdravnika o posebni prehrani. Malformacije se odpravijo s kirurškimi posegi in jih ni mogoče zdraviti s samopomočjo.




.jpg)